McMaster University (2024) announced that a pre-clinical animal (xenograft) model study led by the Singh Lab has discovered that chimeric antigen receptor (CAR) CAR-T cell therapy may be used to treat glioblastoma, a malignant brain cancer. Current treatments for brain cancer were established by Stupp and Colleagues (2005) and consist of maximal surgical resection of the tumour (complete removal of the tumour). This is followed by radiotherapy and chemotherapy (Chouleur et al., 2024).
However, the effectiveness in increasing survival is limited, with a median survival estimated to be 15 months (Aldape et al., 2019). Other limitations include an increased risk of tumour recurrence and resistance to genotoxic therapies (Chokshi et al., 2024). The likely cause of such resistance is the heterogeneous origin of glioblastoma, which may also illustrate intratumor heterogeneity, where both forms pose a challenge and vary with patient (Nicholson and Fine, 2021).
Therefore, the recent development and use of CAR T-cell therapy appear to have made a significant advancement in helping to identify new targets for the therapeutic use of gliomas. Challenges have also been identified in haematological malignancies, where more than 80% paediatric patients with acute lymphoid leukaemia (ALL) were cured with intensive chemotherapy. ALL arises in the B cells, a type of white blood cell. However, there is limited effectiveness for patients with returning cancer/relapse post-chemotherapy or using a stem cell transplant (National Cancer Institute, 2025).
What Are CAR T-Cells?
CARs bind to specific proteins known as antigens present on cancer cells (Cancer Research UK, 2024). Antigens are also found on some normal cells. This strengthens the immune response induced by T cells against cancer cells. The structure of CAR is found in Figure 1. There are four main domains of CAR. The antigen-binding domain, also referred to as the single-chain variable fragments (scFV), is present outside the cell. The scFV is made of artificial antibodies in the laboratory and is used to affect the receptor’s ability to recognise its specific targets. The role of scFV is optimised for binding affinity. This helps minimise toxicity against non-tumour tissues (Park, Maus, and Choi, 2024; National Cancer Institute, 2025; Cancer Research UK, 2024).
The gap, also known as the hinge domain, is a bridge between the antigen-recognising domain and the cell membrane of the T cell (Park, Maus, and Choi, 2024). It helps provide flexibility and length to the CAR. It also facilitates positioning for the CAR to bind to the target antigen (Xiao et al., 2022). The hinge domain is considered a vital point to target solid tumours and haematological malignancies, particularly with interactions, proliferation, and T cell activation (Park, Mause, and Choi, 2024).
The transmembrane domain maintains the stability of CAR on the T cell surface. It connects the inside and outside of the cell and releases cytokines, which help transmit recognition of ligand/signalling molecule (Park, Maus, and Choi, 2024).
The intracellular domain is present inside the cell and is subdivided into co-stimulatory and T-cell activation. This provides a signal once the receptor binds to the antigen of the tumour cell. The signals are transmitted inside the T cells and help maintain their survival by allowing T cells to multiply more and reach millions (National Cancer Institute, 2025; Cancer Research UK, 2024). They are then sent back to the patient as a single infusion (National Cancer Institute, 2025; Cancer Research UK, 2024).
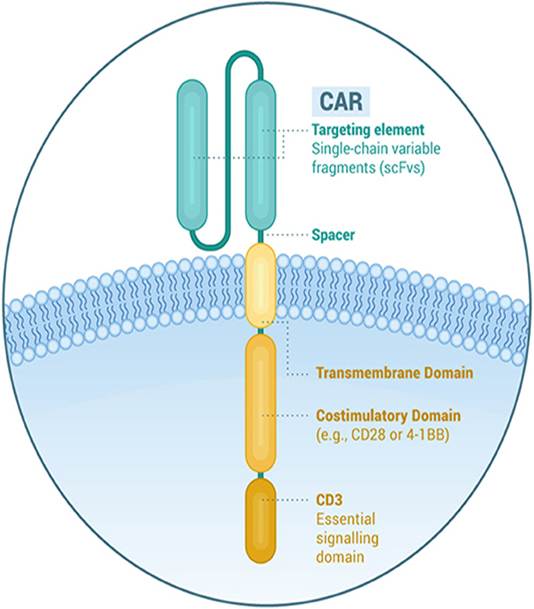
Figure 1: The structure of a T cell
What does the process of CAR T-cell therapy involve?
The entire process of CAR-T cell infusion takes approximately three to five weeks, from the initial collection of blood to the infusion back into the patient. This is illustrated in Figure 3. At first, T cells are collected from the blood by the process of apheresis. The remainder of the blood and fluid re-enter the body via a tube in the other arm. It takes approximately 4 to 6 hours to collect T cells. Nurses and researchers alter T cells to become CAR T cells by adding a gene that causes the production of proteins on the surface called CARs. Allowing the expression of CARs in T cells helps to transduce cells with viral vectors that contain a semi-random integrated transgene into the T cell genome (Zugasti et al., 2025). A transgene is the transfer of a gene from one organism to another organism using artificial techniques.
One of the most recent techniques of gene editing is through CRISPR-mediated editing. CAR is inserted into the T cell receptor (TCR) alpha constant (TRAC). The TRAC locus is found in the alpha chain gene in the T cell receptor. This form of editing helps to suppress the expression of the natural form of TCR, limiting its interference in the recognition of antigens on cancer cells (Zugasto et al., 2025). The presence of TRAC helps to maintain the stability of receptors and recognition of antigens to ultimately eradicate cancer cells. It also helps increase T cell exhaustion, the graft-versus-host effect, and the efficacy of CAR T cell therapy (Zugasti et al., 2025).
Continuous growth of CAR T cells occurs and will then be re-entered into the patient via a drip. The T cells will expand and differentiate with the help of cytokines (Cancer Research UK, 2024). This strengthens the immune system. The CAR-T cell receptors can remain in the body for a long period of time, but on monitoring control over the next two weeks post-treatment.
After this length of time, they are discharged because if the patient has someone with them for the 24 hours. On the contrary, some remain in hospital for the first month after treatment (Cancer Research UK, 2024). Day-care unit, several times a week, may also be organised, and is known as an ambulatory clinic. CAR-T cells that are not in use are frozen.
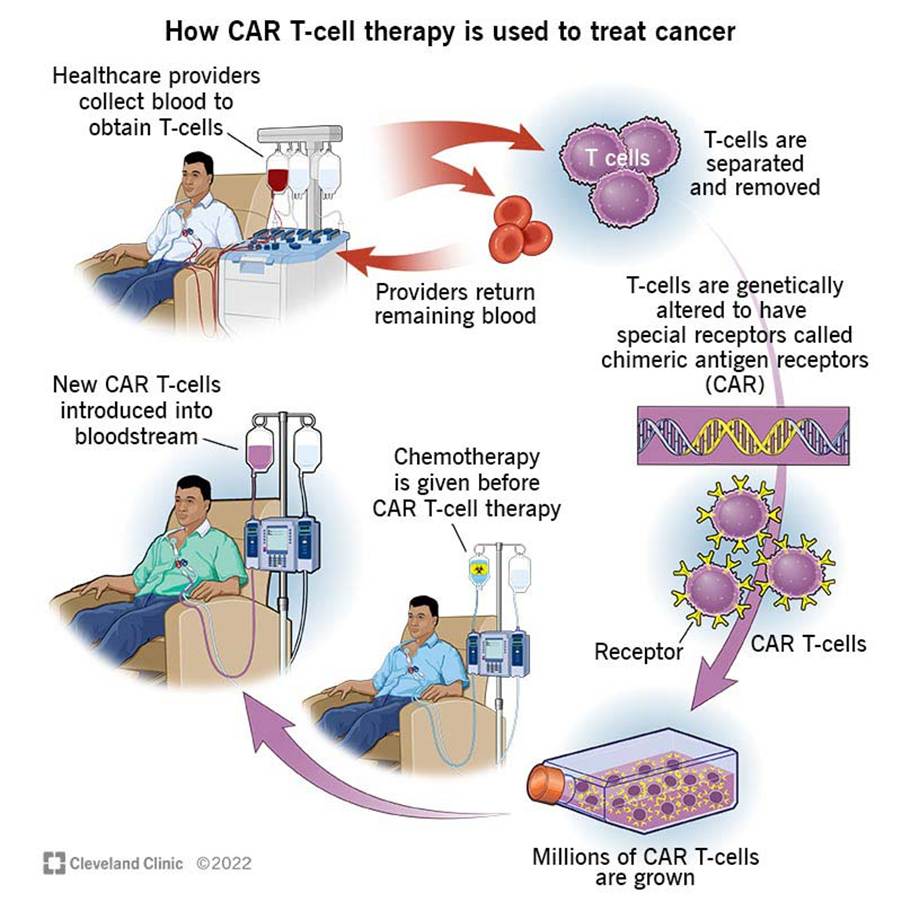
Figure 2: The mechanism of CAR T cell therapy.
Achievements of the Singh lab
The Singh Lab compared three types of cancers: adult glioblastoma, paediatric relapsed medulloblastoma, and adult lung to brain metastasis (McMaster University, 2024).
They undertook large-scale gene editing technology to compare genes before and after. This will help to identify functional gene drivers responsible for increasing the number of mutations. Axonal guidance is vital in neural development. Axons are long chain that carries the electrical message from the cell body receiving the information towards the end of the terminal of the neuron (nerve cells), referred to as the axon terminal. Please see Figure 3. Axons can migrate in response to extracellular guidance molecules on whether to increase or halt activity within the axon (Wadsworth, 2015). This signalling axis can be significantly affected by cancer cells to invade and infiltrate the brain. Please see Figure 4.
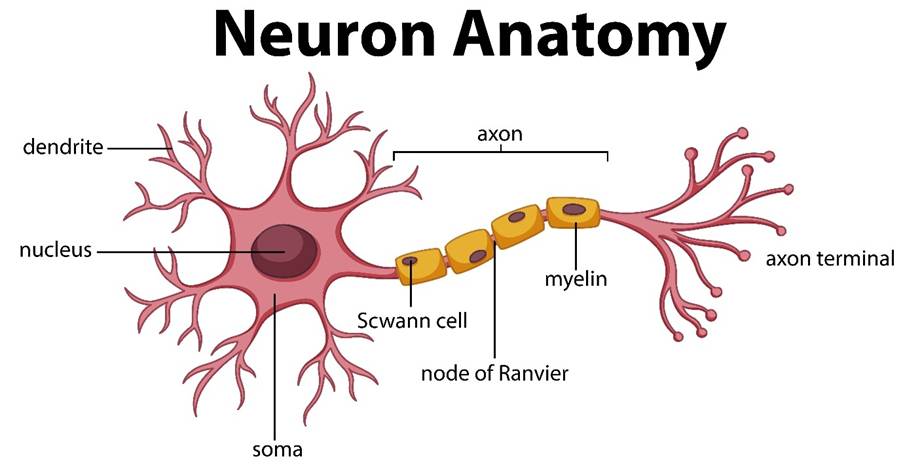
Figure 3: A schematic structure of a neuron (nerve cell)
An Insight Into Axonal Guidance
The tip of the developing neurone is known as the growth cone. The growth cone can help sense the cellular environment and can select its specific pathway (Ichigo, Sugiura, and Kimata, 2013). Some molecules influence how the growth cones perform and are referred to as guidance cues. Attractive guidance cues help with the growth of the action through contact or diffusion. Repulsive guidance cues ensure molecules are kept away from the growth cones. As a result, an inhibitory effect develops simultaneously through contact or diffusion-mediated effects (Ichigo, Sugiura, and Kimata, 2013). This is illustrated in Figure 4.
One of the common model examples used in clinical research to illustrate axonal guidance before the Singh Lab discovery is Caenorhabditis elegans. C. elegans is a free-living nematode due to its simple structure and similarities to human genes. A neurone in C. elegans was found to have been misled to produce a UNC-6 (netrin response). However, many animals have no UNC-6 but have a change in the UNC-6 receptor UNC-40 (DCC) and UNC-53 (NAV2). Neurons would respond to UNC-6, and the direction of axon outgrowth changes (Wadsworth, 2015). For instance, UNC-6 is the UNC-40 receptor and other molecules localised on the membrane where the axon forms (Wadsworth, 2015). Thus, the growth cone helps axons to respond and navigate to targets alongside guidance cues (Wadsworth, 2015).
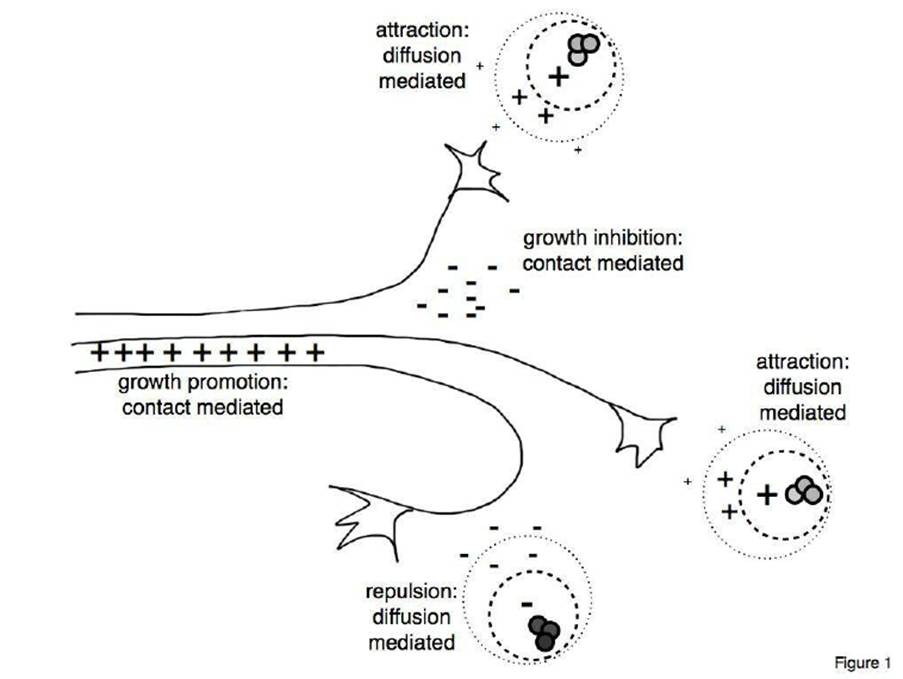
Figure 4: Axonal guidance (Ichigo, Sugiura, and Kimata, 2013)
The Role Of PTP4A2
Amongst the significant observations of the Singh Lab was the finding of the gene protein tyrosine phosphatase 4A2 (PTP4A2). The role of PTP4A2 is primarily in proliferation, forming tumours, and self-renewal. This is a major link in understanding cancer pathogenesis. Axonal guidance would need to be blocked by CAR T cells by targeting Roundabout Guidance Receptor 1 (ROBO1), which guides cells. The effectiveness of the CAR T cell therapy helped to increase the survival rate by two-fold. Chokshi et al. (2024) declared that ROBO1 CAR T cells helped remove 50 to 100% in mouse models. In other words, cells were taken from patients, edited, and placed back with a new role to find ROBO1 on tumour cells. (McMaster University, 2024).
This discovery correlates with findings from Chouleur et al. (2024). One of the genes encoded by PTP4A2 is phosphatase of regenerating liver 2 (PRL2). It can remove a phosphate group (dephosphorylate) from the amino acid residues, tyrosine, serine, and threonine in its target molecule. The target molecule is referred to as the substrate that specifically binds to the enzyme (Chouleur et al., 2024).
PRL2 contributes to cancer pathogenesis when overexpressed in multiple cancers, especially gliomas, and its aggressiveness. The removal of PTP4A2 or its depletion can increase cancer cell death (apoptosis) and induce inflammation-mediated effects in in vivo studies. This slows tumour growth and makes a drastic shift in the tumour microenvironment (TME) from a pathogenic to immunosuppressive state (Chouleur et al., 2024).
Alternatively, PTP4A2 expression does not affect the accumulation of macrophages inside the tumour. Macrophages are a type of white blood cell that has a non-specific immune defence against pathogens. This has been identified by their specific markers, Adgre (F4/80) or Aif1 (IBA1), where it was not differentially expressed (Chouleur et al., 2024).
The TME that surrounds solid tumours and can cause infiltration of multiple immune cells to induce an anti-tumour response, namely, regulatory T cells and myeloid-derived suppressor cells, cytokines, growth factors, and tumour-signalling molecules (Gabrusiewicz et al., 2011).
The T cell-mediated effects are evaded by tumour cells using checkpoint molecules, namely programmed death ligand 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) (Park, Maus and Choi, 2024).
Another mechanism that facilitates tumour-associated impairment of the immune response in gliomas is hypoxia through the increased expression of hypoxia-inducible factor 1-alpha (HIF1-α) (Kumar and Gabrilovich, 2014). This illustrates that the brain interconnects with the immune system, where immune cells can bypass multiple protective barriers. For example, the blood-brain barrier and the blood-cerebrospinal fluid barrier, where T cells can readily enter the tight junctions (Park, Maus, and Choic, 2024). Please see Figure 5.
The prominent role of the blood-brain barrier is to protect the brain from harmful chemicals and toxins by applying an inclusive regulatory mechanism of ions, large molecules, and immune cells into the CNS and maintaining brain homeostasis. The blood-cerebrospinal fluid barriers limit communication between the different anatomical structures of the brain and are formed by the choroid plexus epithelium (Park, Maus, and Choi, 2024).
Moreover, it is important to state that there are several isoforms of PTP4A. Yu et al. (2023) conducted a pan-cancer analysis of its normal and aberrant genetic expression and function. Amplification was the main form of alteration, where PTP4A3 is mostly upregulated. In descending order, this is followed by PTP4A2, which had a generic upregulation (Yu et al., 2023).
Further investigations are needed for PTP4A1 due to the inconsistency of results despite the same cancer and analysis being conducted across different datasets. There was an independent relation between the genetic and protein expression of PTP4As. This highlights the complexity of their regulatory mechanisms (Yu et al., 2023).
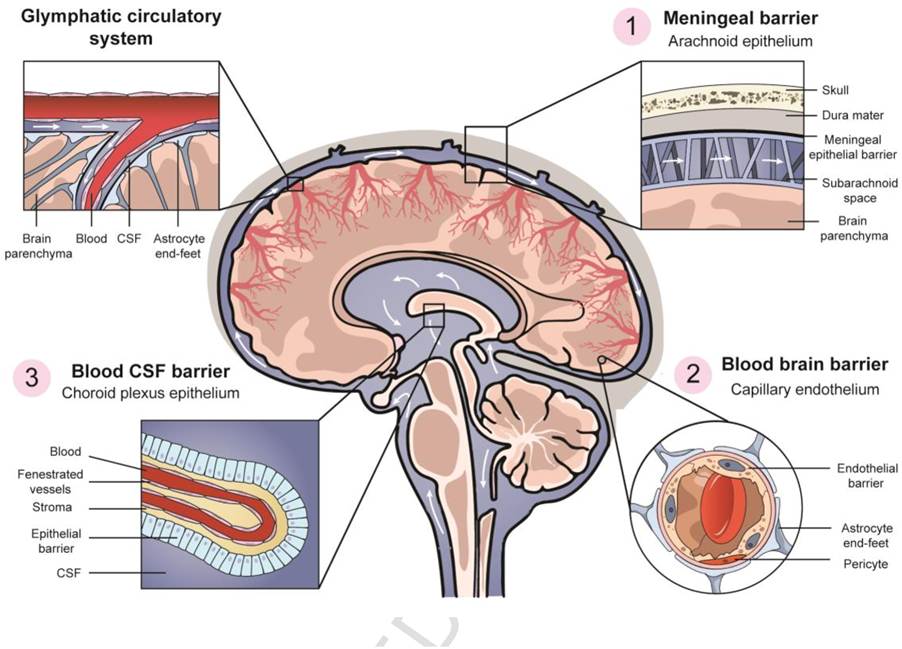
Figure 5: The four main protective barriers in the brain.
Other Genes Found In Glioma
Besides PTP4A2 and ROBO1, there are other antigens that are overexpressed in gliomas. They are considered CAR T-cell therapeutic targets and other immunotherapeutic modalities. B7 homolog 3 protein (B7-H3), also known as CD276 are type 1 transmembrane immune checkpoint protein (Tang et al., 2019).
Epidermal Growth Factor Receptor (EGFR) type 3 is a transmembrane protein and was found to be amplified in 30% of glioma patients and not in any healthy tissues (Wikstrand et al., 1995).
EphA2 receptor protein has been found in 90% of glioma cases and low levels were depicted in non-cancerous tissues, where it facilitates migration, differentiation, growth, and modulates the signal transduction pathway (van der Geer, Hunter, and Lindberg, 1994; Wykosky et al., 2005).
Interleukin-13 (IL-13Rα2) is overexpressed in 50 to 80% of gliomas and has very low levels untraceable to measure IL-4 in normal tissues. In normal states, IL-54 co-regulates 13Rα2 immune response.
In contrast, gangliosides (GD2) consist of glycosphingolipids whose structure has sugars and fats. GD2 is widely expressed in normal and glioma tissues. However, the percentage of gangliosides in normal neural tissues in the central and peripheral nervous system is found to be less than 4%. Experimental studies in murine and cellular models have found that CAR T-cells have potent cytotoxicity against gliomas (Golinelli et al., 2020; Prapa et al., 2015).
A similar percentage, up to 80%, was found for the receptor tyrosine kinase HER2 as IL-13Rα2. However, HER2 has a dual effect. Morgan et al. (2010) discovered that CAR-T cells reported to induce fatal toxicity and multiorgan system failure in patients with colon cancer that metastasised to the lungs and liver. On the contrary, second-generation CAR T-cell therapy was deemed safe and did not cause dose-limiting toxicities (Ahmed et al., 2017). This highlights that the design, mode of action of CAR-T cell therapy, and infusion conditions contribute to the aftermath.
CAR-T Cell Toxicity
Our current understanding of how CAR-T cell-associated neurotoxicity arises requires dire improvement. The most common CAR-T cell toxicity is of two types: cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS). Both appear predominantly in the treatment of blood cancers and in the absence of CNS tumours in hours or days after CAR T-cell therapy, and in some cases, late onset (Park, Maus, and Choi, 2024). Inflammation also increases and contributes to pathogenesis, where levels of cytokines, tumour necrosis factor-alpha TNF-α, interleukin 6, and interferon-gamma (IFN-γ) are raised upon infusion. Both cytokines activate additional immune cells, monocytes, and macrophages to produce more cytokines, for example, interleukin 1, interleukin ,6 and inducible nitric oxide synthase. CRS is characterised by low blood pressure, fast heartbeat, hypoxia, and fever, and in severe cases, multiorgan failure.
ICANS occurs in 40% of patients undergoing CAR T cell therapy (Gust et al., 2017). It can disrupt the blood-brain barrier via cytokine release and increase permeability. This induces neuroinflammation, cerebral oedema, and thrombotic microangiopathy. Thrombotic microangiopathy where small blood vessels and the lining (endothelium) are injured. Cerebral oedema is the build-up of fluid in the brain. Additional symptoms may arise, such as seizures, confusion, deliriu,m and difficulty in language and speech (aphasia) (Santomasso et al., 2018).
It has been speculated that localised tumour inflammation-associated neurotoxicity (TIAN) is dependent on where the tumour is in the brain and the surrounding regions, where symptoms may arise within days and weeks after CAR T cell therapy. Its effects are concurrent with CRC.
Type 1 TIAN is mechanistically linked to causing headache, neurocognitive deficits, and consciousness. On the contrary, TIAN type 2 causes seizures, confusion, and mental status (Park, Maus, and Choi, 2024). To differentiate, some markers appear elevated but evolve rapidly per patient. This highlights the need for additional diagnostic testing for accuracy.
Other substantial challenges in CAR T cell therapy are finding an antigen that is solely present on cancer cells. An immunosuppressive state in tumours can cause CAR T cells to malfunction. Tumour heterogeneity may vary per patient and within the same patient, where antigens appear at low levels for CAR T cells to work (National Cancer Institute, 2025).
Furthermore, another technique in process, which is collecting T cells from healthy donors rather than disease patients. This is known as allogenic blood transplantation and can be done in advance, rather than a waiting period of weeks for patients. Conversely, it requires genetic engineering to prevent the immune system from recognising T cells as foreign. It is estimated that patients who respond well to CAR T-cell therapy have an additional two years without the need for chemotherapy (National Cancer Institute, 2025).
Overall, there have been immense efforts to drive gene and protein engineering in CAR T-cell development. Researchers are continuing to develop safe, precise CAR T cells for brain cancers with lower side effects, and Singh’s intervention is vital for better progress.
References
Ahmed, N., Brawley, V., Hegde, M., Bielamowicz, K., Kalra, M., Landi, D., Robertson, C., Gray, T.L., Diouf, O., Wakefield, A., Ghazi, A., Gerken, C., Yi, Z., Ashoori, A., Wu, M.-F., Liu, H., Rooney, C., Dotti, G., Gee, A., and Su, J. (2017). HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma. JAMA Oncology, 3(8), p.1094. doi: https://doi.org/10.1001/jamaoncol.2017.0184.
Aldape, K., Brindle, K.M., Chesler, L., Chopra, R., Gajjar, A., Gilbert, M.R., Gottardo, N., Gutmann, D.H., Hargrave, D., Holland, E.C., Jones, D.T.W., Joyce, J.A., Kearns, P., Kieran, M.W., Mellinghoff, I.K., Merchant, M., Pfister, S.M., Pollard, S.M., Ramaswamy, V. and Rich, J.N. (2019). Challenges to curing primary brain tumours. Nature Reviews Clinical Oncology, [online] 16(8), pp.509–520. doi: https://doi.org/10.1038/s41571-019-0177-5.
Cancer Research UK (2024) CAR T-cell therapy. Available at: https://www.cancerresearchuk.org/about-cancer/treatment/targeted-cancer-drugs-immunotherapy/CAR-T-cell-therapy (Accessed: 11th November 2025)
Chokshi, C.R., Shaikh, M.V., Brakel, B., Rossotti, M.A., Tieu, D., Maich, W., Anand, A., Chafe, S.C., Zhai, K., Suk, Y., Kieliszek, A.M., Miletic, P., Mikolajewicz, N., Chen, D., McNicol, J.D., Chan, K., Tong, A.H.Y., Kuhlmann, L., Liu, L., and Alizada, Z. (2024). Targeting axonal guidance dependencies in glioblastoma with ROBO1 CAR T cells. Nature Medicine, pp.1–11. doi: https://doi.org/10.1038/s41591-024-03138-9.
Chouleur, T., Emanuelli, A., Wilfried Souleyreau, Marie-Alix Derieppe, Teo Leboucq, Hardy, S., Mathivet, T., Tremblay, M.L. and Bikfalvi, A. (2024). PTP4A2 Promotes Glioblastoma Progression and Macrophage Polarization under Microenvironmental Pressure. Cancer Research Communications, 4(7), pp.1702–1714. doi: https://doi.org/10.1158/2767-9764.crc-23-0334.
Gabrusiewicz, K., Ellert-Miklaszewska, A., Lipko, M., Sielska, M., Frankowska, M. and Kaminska, B. (2011). Characteristics of the Alternative Phenotype of Microglia/Macrophages and its Modulation in Experimental Gliomas. PLoS ONE, 6(8), p.e23902. doi: https://doi.org/10.1371/journal.pone.0023902.
Golinelli, G., Grisendi, G., Prapa, M., Bestagno, M., Spano, C., Rossignoli, F., Bambi, F., Sardi, I., Cellini, M., Horwitz, E.M., Feletti, A., Pavesi, G. and Dominici, M. (2018). Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Therapy, 27(7-8), pp.558–570. doi: https://doi.org/10.1038/s41417-018-0062-x.
Gust, J., Hay, K.A., Hanafi, L.-A., Li, D., Myerson, D., Gonzalez-Cuyar, L.F., Yeung, C., Liles, W.C., Wurfel, M., Lopez, J.A., Chen, J., Chung, D., Harju-Baker, S., Özpolat, T., Fink, K.R., Riddell, S.R., Maloney, D.G. and Turtle, C.J. (2017). Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discovery, [online] 7(12), pp.1404–1419. doi: https://doi.org/10.1158/2159-8290.CD-17-0698.
Ichijo, H., Sugiura, N., and Kimata, K. (2013). Application of Chondroitin Sulfate Derivatives for Understanding Axonal Guidance in the Nervous System during Development. Polymers, 5(1), pp.254–268. doi: https://doi.org/10.3390/polym5010254.
Kumar, V. & Gabrilovich, D. I. (2014) Hypoxia‐inducible factors in regulation of immune responses in tumour microenvironment. Immunology 143, pp. 512–519.
McMaster University (2024) Brain Cancer Breakthrough: New Therapy Destroys Glioblastoma in Recently Unveiled Pathway Available at: https://scitechdaily.com/brain-cancer-breakthrough-new-therapy-destroys-glioblastoma-in-recently-unveiled-pathway/#:~:text=A%20groundbreaking%20study%20introduces%20a%20therapy%20that%20uses,survival%20times%20and%20eradicating%20tumors%20in%20preclinical%20models (Accessed: 7th November 2025)
Morgan, R.A., Yang, J.C., Kitano, M., Dudley, M.E., Laurencot, C.M. and Rosenberg, S.A. (2010). Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Molecular Therapy, 18(4), pp.843–851. doi: https://doi.org/10.1038/mt.2010.24.
National Cancer Institute (2025) CAR T Cells: Engineering Patients’ Immune Cells to Treat Their Cancers Available at: https://www.cancer.gov/about-cancer/treatment/research/car-t-cells (Accessed: 11th November 2025)
Nicholson, J.G. and Fine, H.A. (2021). Diffuse Glioma Heterogeneity and Its Therapeutic Implications. Cancer Discovery, 11(3), pp.575–590. doi: https://doi.org/10.1158/2159-8290.cd-20-1474.
Park, S., Maus, M.V. and Choi, B.D. (2024). CAR-T cell therapy for the treatment of adult high-grade gliomas. npj Precision Oncology, [online] 8(1). doi: https://doi.org/10.1038/s41698-024-00753-0.
Prapa, M., Caldrer, S., Spano, C., Bestagno, M., Golinelli, G., Grisendi, G., Petrachi, T., Conte, P., Horwitz, E.M., Campana, D., Paolucci, P. and Dominici, M. (2015). A novel anti-GD2/4-1BB chimeric antigen receptor triggers neuroblastoma cell killing. Oncotarget, 6(28), pp.24884–24894. doi: https://doi.org/10.18632/oncotarget.4670.
Santomasso, B.D., Park, J.H., Salloum, D., Riviere, I., Flynn, J., Mead, E., Halton, E., Wang, X., Senechal, B., Purdon, T., Cross, J.R., Liu, H., Vachha, B., Chen, X., DeAngelis, L.M., Li, D., Bernal, Y., Gonen, M., Wendel, H.-G. and Sadelain, M. (2018). Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discovery, [online] 8(8), pp.958–971. doi: https://doi.org/10.1158/2159-8290.cd-17-1319.
Stupp, R., Mason, W.P., van den Bent, M.J., Weller, M., Fisher, B., Taphoorn, M.J.B., Belanger, K., Brandes, A.A., Marosi, C., Bogdahn, U., Curschmann, J., Janzer, R.C., Ludwin, S.K., Gorlia, T., Allgeier, A., Lacombe, D., Cairncross, J.G., Eisenhauer, E. and Mirimanoff, R.O. (2005). Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. New England Journal of Medicine, [online] 352(10), pp.987–996. doi: https://doi.org/10.1056/nejmoa043330.
Tang, X., Zhao, S., Zhang, Y., Wang, Y., Zhang, Z., Yang, M., Zhu, Y., Zhang, G., Guo, G., Tong, A., and Zhou, L. (2019). B7-H3 as a Novel CAR-T Therapeutic Target for Glioblastoma. Molecular Therapy Oncolytics, [online] 14, pp.279–287. doi: https://doi.org/10.1016/j.omto.2019.07.002.
van der Geer, P., Hunter, T., and Lindberg, R.A. (1994). Receptor Protein-Tyrosine Kinases and Their Signal Transduction Pathways. Annual Review of Cell Biology, 10(1), pp.251–337. doi: https://doi.org/10.1146/annurev.cb.10.110194.001343.
Wadsworth, W. (2015). Understanding axon guidance: attraction, repulsion, and statistical physics. Neural Regeneration Research, 10(2), p.176. doi: https://doi.org/10.4103/1673-5374.152360.
Wikstrand, C.J., Hale, L.P., Batra, S.K., Hill, M.L., Humphrey, P.A., Kurpad, S.N., McLendon, R.E., Moscatello, D., Pegram, C.N., Reist, C.J., Traweek, S.T., Wong, A.J., Zalutsky, M.R. and Bigner, D.D. (1995). Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. PubMed, 55(14), pp.3140–8.
Wykosky, J., Gibo, D.M., Stanton, C. and Debinski, W. (2005). EphA2 as a Novel Molecular Marker and Target in Glioblastoma Multiforme. Molecular Cancer Research, 3(10), pp.541–551. doi: https://doi.org/10.1158/1541-7786.mcr-05-0056.
Xiao, Q., Zhang, X., Tu, L., Cao, J., Hinrichs, C.S. and Su, X. (2022). Size-dependent activation of CAR-T cells. Science Immunology, 7(74). doi: https://doi.org/10.1126/sciimmunol.abl3995.
Yu, M., Lin, C., and Wei, M. (2023). A pan-cancer analysis of oncogenic protein tyrosine phosphatase subfamily PTP4As. Journal of Holistic Integrative Pharmacy, [online] 4(2), pp.185–198. doi: https://doi.org/10.1016/j.jhip.2023.07.001.
Zugasti, I., Espinosa-Aroca, L., Fidyt, K., Mulens-Arias, V., Diaz-Beya, M., Juan, M., Urbano-Ispizua, Á., Esteve, J., Velasco-Hernandez, T., and Menéndez, P. (2025). CAR-T cell therapy for cancer: current challenges and future directions. Signal Transduction and Targeted Therapy, [online] 10(1). doi: https://doi.org/10.1038/s41392-025-02269-w.

Leave a comment